
北京大学深圳研究生院研究员、科学智能中心主任陈语谦与新加坡国立大学合作,在Nature Communications发表了题为“Scalable crystal structure relaxation using an iteration-free deep generative model with uncertainty quantification”的研究论文。该论文提出了一种单步晶体结构优化方法,避免了在使用第一性原理计算进行晶体结构优化时的迭代过程,为大规模晶体结构优化和高通量计算提供了高效的解决方案。

论文封面
原子或晶体结构优化在计算化学、计算物理和计算材料科学等领域具有广泛的应用。结构优化的主要目标是找到最低能量状态,也称为基态。基态结构是计算并预测其物理和化学性质的基础。该过程对表面化学反应、晶体缺陷调控以及药物设计等应用至关重要。
传统上,晶体结构优化常采用第一性原理计算方法,例如密度泛函理论(DFT)。DFT通过迭代计算电子密度和总能量,调整原子位置以最小化系统能量,从而实现晶体结构优化。尽管基于DFT的结构优化方法精度较高,但其高计算需求和较差的可扩展性限制了其在大规模晶体结构优化和高通量计算场景中的实用性。因此,在这些应用中,迫切需要一种计算量小、可扩展性强、通用性广的晶体结构优化方法。
在这项研究中,作者开发了一种机器学习模型——DeepRelax。DeepRelax通过单步计算,直接预测出比初始结构更稳定的结构,避免了DFT中需要迭代计算电子密度和总能量的计算瓶颈。进一步使用DFT对DeepRelax预测的结构进行优化,可以实现快速收敛。在使用单个A6000 GPU进行计算时,DeepRelax仅需几百毫秒便能完成一个晶体结构的优化工作,与发表在Nature Computational Science上的M3GNet相比,速度提升了两个数量级。此外,DeepRelax还具备并行结构优化的能力,这一特点使其在高通量材料筛选和计算中的应用价值得到了显著提升。
为了验证DeepRelax 的通用性,论文对其进行了全面的验证,包括三维材料和二维材料在内的多个数据集,如X-Mn-O氧化物体系(X代表 Mg、Ca、Sr and Ba)、Materials Project、C2DB、缺陷结构、范德瓦尔斯晶体。实验结果表明,在所有体系中,DeepRelax预测的原子坐标以及晶格常数,相比未优化的结构均有明显的提升。对DeepRelax预测的结构进行DFT验证,结果表明这些结构的能量显著低于未优化的结构。此外,将DeepRelax预测的结构作为初始结构进行DFT优化,可以显著减少达到基态结构所需的迭代步数。这进一步证明了DeepRelax在加速DFT计算、提高计算效率方面的巨大潜力。
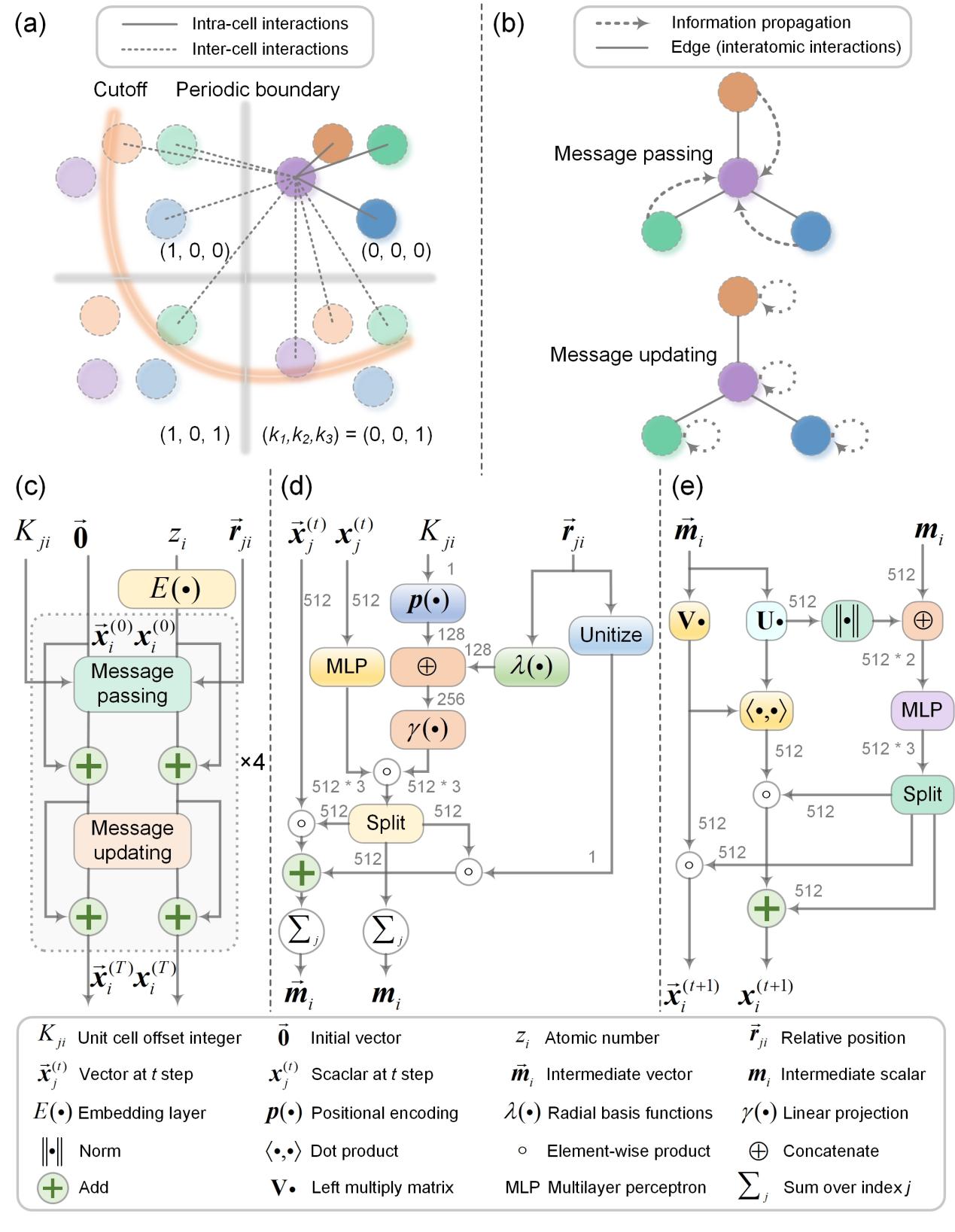
图1 DeepRelax模型架构
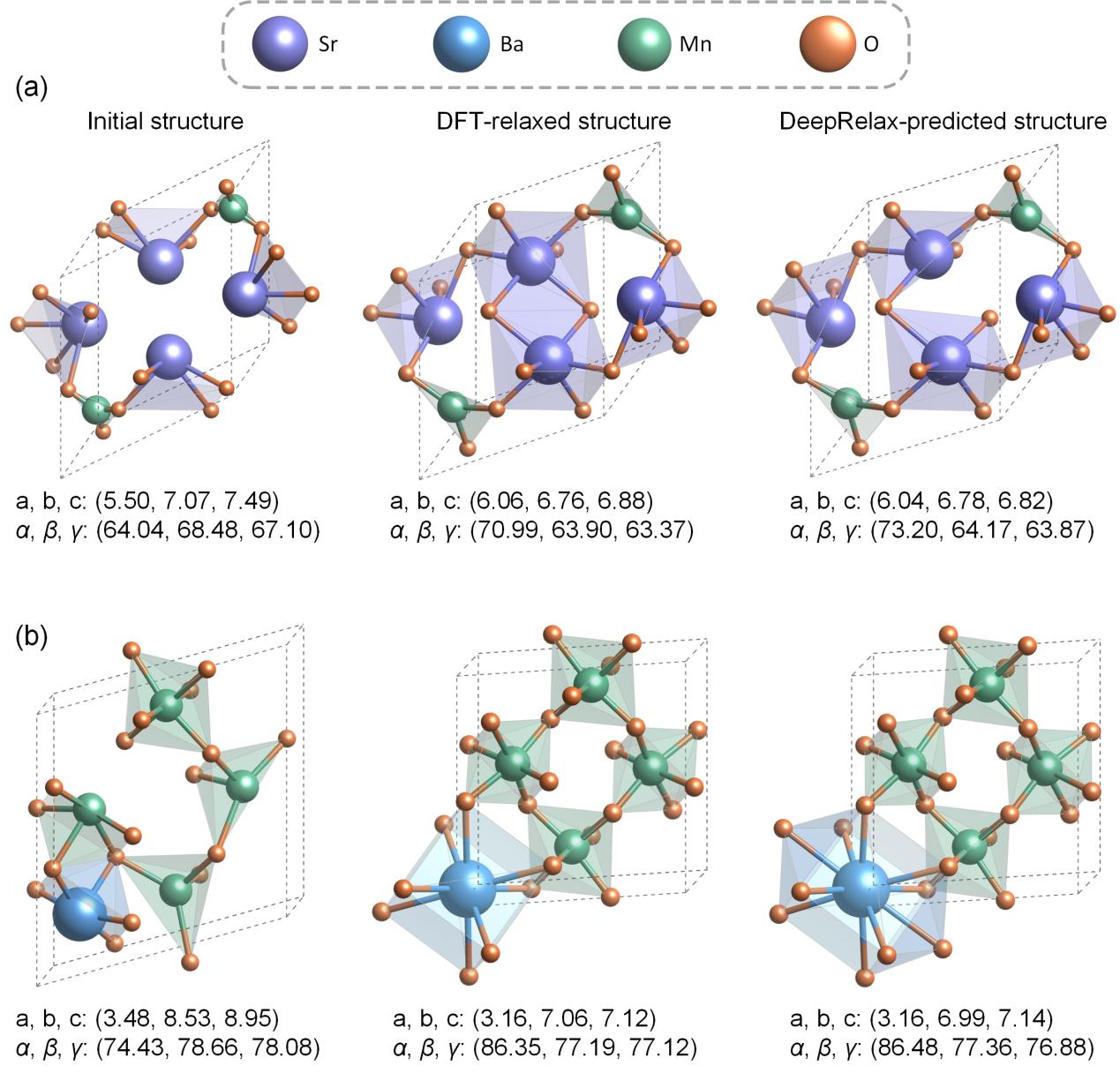
图2 使用DeepRelax进行结构优化的例子
陈语谦与新加坡国立大学Lei Shen教授为共同通讯作者;中山大学博士生、北京大学深圳研究生院研究助理杨梓铎与新加坡国立大学博士生赵艺明为共同第一作者。
文章来源:北京大学







